load library
library(rtracklayer)
library(tidyverse)
library(ChIPpeakAnno)
library(ggsci)
load peak files
From “The ChIPpeakAnno user’s guide”
bed <- system.file("extdata", "MACS_output.bed", package="ChIPpeakAnno")
gr1 <- toGRanges(bed, format="BED", header=FALSE)
gff <- system.file("extdata", "GFF_peaks.gff", package="ChIPpeakAnno")
gr2 <- toGRanges(gff, format="GFF", header=FALSE, skip=3)
## If you are importing files downloaded from ensembl,
## it will be better to import the files into a TxDb object,
## and then convert to GRanges by toGRanges. Here is the sample code:
## library(GenomicFeatures)
## txdb <- makeTxDbFromGFF('/config/binaries/R/4.2.0/R_libraries/ChIPpeakAnno/extdata/GFF_peaks.gff')
## anno <- toGRanges(txdb, format='gene')
ol <- findOverlapsOfPeaks(gr1, gr2)
## duplicated or NA names found.
## Rename all the names by numbers.
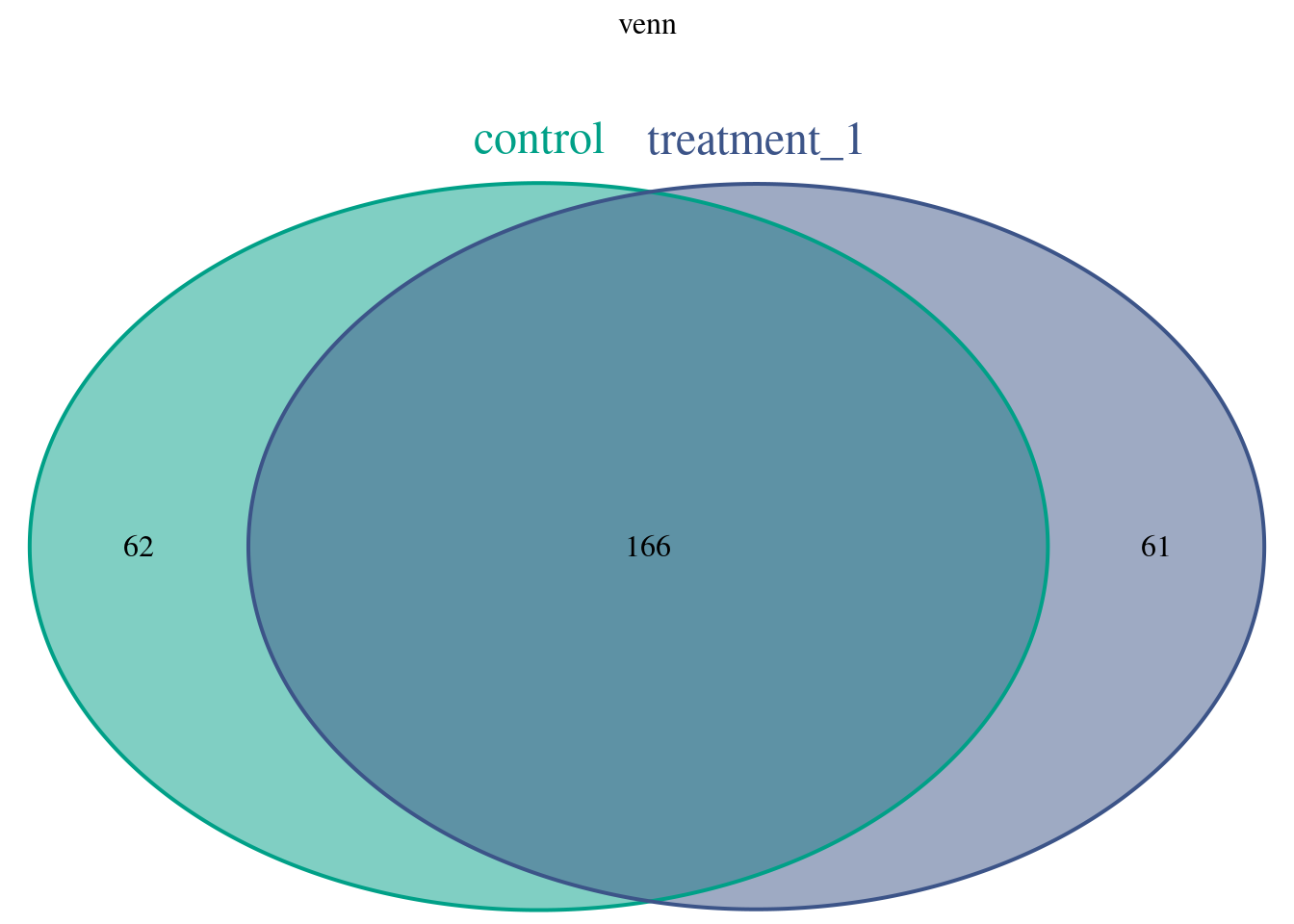
make venn diagram
col_npg5 <- c(ggsci::pal_npg()(10)[c(3,4,9,1,2)])
# don't write log file for VennDiagram
futile.logger::flog.threshold(futile.logger::ERROR, name = "VennDiagramLogger")
makeVennDiagram(ol,
fill=col_npg5[1:2], # circle fill color
col=col_npg5[1:2], #circle border color
#connectedPeaks = "merge", connectedPeaks = "keepAll", min keepFirstListConsistent
cat.col=col_npg5[1:2], connectedPeaks = "merge", scaled=T, by = "region", main = "venn", NameOfPeaks = c("control","treatment_1"), cat.cex = 1.5, cat.pos = c(0, 0))